Molecular Materials and Transport
Density Functional Theory for Transport Applications
The application of density functional theory (DFT) to transport problems has a long history in the field of Molecular Electronics. The DFT-technology is so important, because it can connect the molecular structure to its observable properties, which often are sensitive to changing just a single atom. DFT-transport theories rely upon two central ingredients. Future improvements require work in both directions and we are active there.
.") Nonequilibrium Green's Function Approach: AitransS
Nonequilibrium Green's Function Approach: AitransS
The results of any first principles calculation need to be converted into transport information, like transmission functions, charge and heat conductance etc. To this end we employ the nonequilibrium Green's functions approach. Our particular methodology is implemented in a transport module -- AitransS-- that currently is running with standard DFT-codes TURBOMOLE and FHI-aims .
.")
Improvements and extensions of this transport module are continuously under way. This includes the calculation of local current densities, orbital magnetism or spin-orbit torques out of equilibrium.
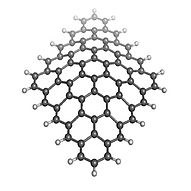
Current pattern in large functionalized graphene flakes
Using our transport tool, we also investigate the effect of functionalization on conductance and the current pattern through large mesoscopic flakes. Usually these currents vary greatly on microscopic length scales. Our DFT approach includes effects like lattice distortion, e.g. strain, and cross-talk between different impurities.
Current flows in loops in graphene sheets: find more in our Physical Review Letters paper.
Incommensurate oscillations in the excitation gaps of molecular wires
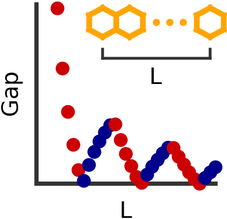 The optical gaps of oligoacenes show incommensurate oscillations as a function of length. | Basic oligacenes (benzene, naphthalene, anthracene, ...) are well known molecules, which consist of linearly fused benzene rings. We have shown that the honey-comb-like arrangement of carbon bonds leads to a surprising periodicity of the properties of oligoacenes, as we keep adding more and more rings. We argue that this is a natural consequence of the band structure of the infinite chain (polyacene), having a relativistic dispersion close to the Fermi points. |
Webmaster Evers - 24.06.2021 17:52