

P4-ACTIVATION
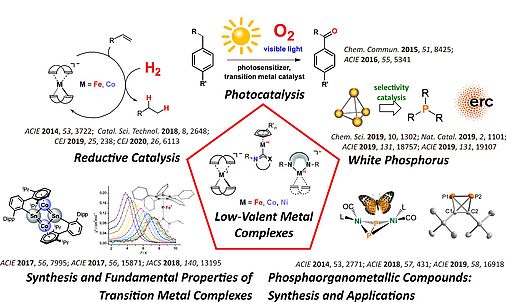
White Phosphorus Activation
European Research Council (ERC) Consolidator Grant FunctionalP4:
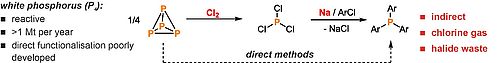
Organophosphorus compounds (OPCs) are a very important and industrially relevant class of substances with a wide range of applications. For example, OPCs are used in organic synthesis, catalysis, as flame retardants and photoinitiators. So far, the vast majority of these valuable and useful compounds have been prepared in atom-inefficient, multi-stage processes in which the white phosphorus (P4) is first oxidized with toxic chlorine gas to chlorinated intermediates (especially PCl3). During the subsequent product formation through salt metathesis, large amounts of highly undesirable chlorine-containing waste are produced. Although more efficient and environmentally friendly synthetic methods are urgently needed, direct, catalytic methods for the conversion of P4 into OPCs are so far almost unknown.
.
.
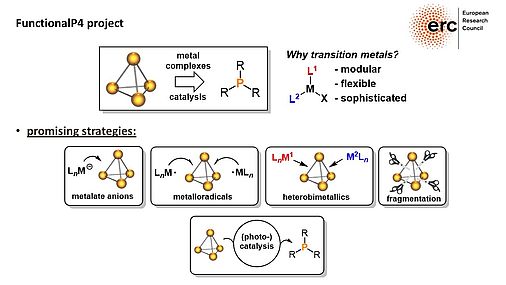
The project FunctionalP4 explores new methods for the activation and functionalization of white phosphorus. The metal-mediated stepwise transformation of P4 into organophosphorus compounds is a key objective. Novel transition metal compounds are designed and synthesized, which can generate reactive phosphorus units. The concept of heterobimetallic P4 activation, where two electronically different metal complexes interact with P4 cooperatively, is introduced for this purpose. Reactions of the phosphorus fragments in these new, reactive complexes with electrophiles will produce novel, fundamentally interesting organophosphorus compounds avoiding chlorinated intermediates. Catalytic methods for P4 functionalization are currently unknown, and developing such methods using transition metal and photoredox catalysts is an additional objective of this proposal.
By providing novel synthetically useful and even catalytic procedures for converting P4 into organophosphorus compounds, this project significantly contributes to the development of phosphorus chemistry and more sustainable synthesis methods.
Selected recent publications: Chem. Sci. 2019, 10, 1302–1308; Angew. Chem. Int. Ed. 2019, 58, 18931–18936; Nat. Catal. 2019, 2, 1101–1106; Angew. Chem. Int. Ed. 2020, 132, 14252–14257; Nat. Chem. 2021, 13, 458–464.
REDUCTIVE CATALYSIS
DFG Research Training Group 2620 Ion Pairs in Reaction (project P10)
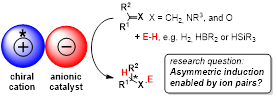
Chiral Cation-Directed Hydrofunctionalization Catalysis
Our project within the graduate school Ion Pairs in Reaction harnesses ion-pairing of chiral organic cations with (achiral) anionic catalysts for asymmetric hydrofunctionalisation reactions. A variety of suitable anionic catalysts are studied. PhD students will receive training in the synthesis of organic and organometallic molecules, catalytic studies, and mechanistic investigations.
Further information on the RTG can be found through the following link: https://www.uni-regensburg.de/chemistry-pharmacy/grk-2620/home/index.html
Applications (single .pdf document) should typically include a letter of motivation, CV, an academic transcript of records, and contact information of two references, preferable in English. Prospective PhD students interested in the Researcht Training Group Ion Pairs in Reaction should apply exclusively to rtg-ion-pairs@uni-regensburg.de
.
Hydrogenations
Catalytic hydrogenations constitute one of the most important operations for the synthesis of fine chemicals and pharmaceuticals. In collaboration with our colleage Prof. Axel Jacobi von Wangelin (UR) we recently discovered that bis(anthracene)cobaltate 1 and -ferrate 2 are potent precatalysts for the hydrogenation of alkenes, ketones and imines under mild conditions. The active catalyst appears to be stabilized by π-coordination of the substrate in the case of alkene hydrogenations, while the hydrogenation of ketones and imines likely involves the formation of catalytically active cobalt particles.
The goal of this project is to further explore the scope and mechanism of arene- and alkene-based catalysts. In order to identify the nature of the catalytically active species, we pursue catalyst poisoning experiments, synthesize model complexes and perform NMR monitoring and ESI-MS studies (the latter in cooperation with the group of Konrad Koszinowski at the University of Göttingen).
.
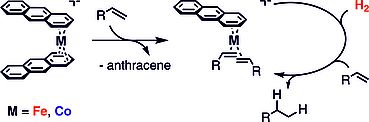
Angew. Chem. Int. Ed. 2014, 53, 3722-3726.
.
Cross-Couplings
Well defined, low-valent anthracene iron(-I) and cobalt(-I)complexes are competent precatalysts for cross-coupling reactions. Other organoiron and -cobalt complexes that we investigated show little to no activity. The oxidation state of the metal in the catalyst precursors has little bearing on the catalytic performance, but a labile coordination environment seems crucial for high catalytic activities.
.
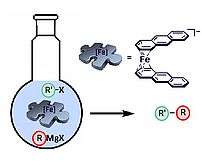
ChemCatChem 2011, 3, 1572-1577; Chem. Sci. 2013, 4, 776-784.
.
PHOTOCATALYSIS
Collaborative Research Center (CRC) 325 Assembly-Controlled Photocatalysis (project A4)
In collaboration with research groups at Regensburg, Munich and Leipzig, we develop photoredox reactions catalyzed by 3d transition metal complexes. Use of substrate-catalyst preassembly offers the chance to overcome some of the key shortcomings of current 3d metal photocatalysts, specifically the extremely short excited state lifetimes of most 3d metal species. Building upon promising preliminary work, this project will investigate ‘outer sphere’ interactions, in which the substrate interacts with the ligand sphere of the metal (e.g. through π-π interactions with heterocyclic ligands or hydrogen bonding to pendant hydrogen bond donors), and ‘inner sphere’ interactions, in which the substrate binds strongly or weakly at the metal atom. In both cases, coordination chemical studies will determine the optimal design principles for the envisaged photocatalysts, alongside analysis of their electronic structures. To guide the development of this project it will be crucial to understand the specific interactions that exist between substrates and catalysts both in the ground and excited states. This fundamental knowledge will then permit the targeted development of applications in organic synthesis, which harness the specific advantages of the developed catalysts in comparison with precious metal compounds.
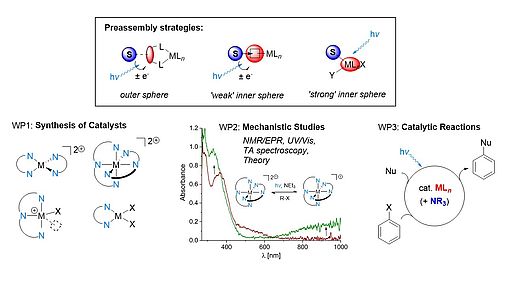
Further information on the CRC can be found through the following link: https://go.ur.de/crc325
Prospective PhD students interested in the CRC Assembly-Controlled Photocatalysis should apply exclusively to apply-crc325@ur.de. Applications (single .pdf document) should typically include a letter of motivation, CV, an academic transcript of records, and contact information of two references, preferably in English.
.
Photocatalytic Oxygenations via Coupled Redox Photocatalysis
The selective oxidation of alkenes and aliphatic hydrocarbons represents a major challenge in modern chemistry. In nature, these oxidations are performed successfully under mild conditions by metalloenzymes such as cytochrome P450 and non-heme based oxygenases. In the lab, a plethora of successful transition metal-based catalysts have been developed. However, only few catalysts are able to use atmospheric oxygen as the terminal oxidant, and these rare examples still show poor selectivities. Moreover, few examples for photocatalytic oxyfunctionalizations have been described to date.
In this project, we develop new photocatalytic methods for the selective oxyfunctionalization of hydrocarbons. Preferably, atmospheric oxygen is used as the oxidant. The investigations are conducted in the framework of the research training group "Chemical Photocatalysis" (GRK 1626).
.
.
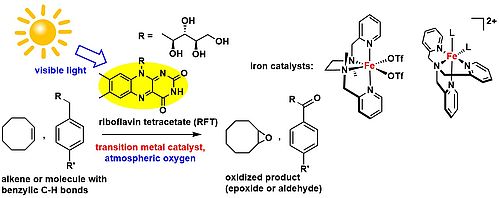
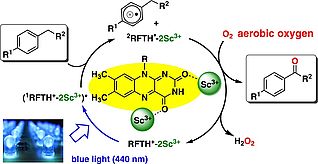
Photocatalytic Benzylic C-H Bond Oxidation with a Flavin Scandium Complex
The use of flavins as photoreceptors and redox cofactors by nature has inspired the development of synthetic flavin analogues as photocatalysts. Riboflavin tetraacetate (RFT) is a prominent example. The photocatalytic C-H bond oxidation of alkyl benzenes to the corresponding aldehydes is one of its particularly intriguing applications. However, the substrate scope was previously limited to substrates with strongly electron-donating substituents on the arene ring. Fukuzumi et al. reported that the reduction potential of RFT can be increased by complex formation with Lewis acidic scandium(III) ions. In this project we have now found that this RFT/Sc(OTf)3 system allows the efficient catalytic oxidation of benzylic C-H bonds by aerobic oxygen.
.
Chem. Commun. 2015, 51, 8425-8428.
.
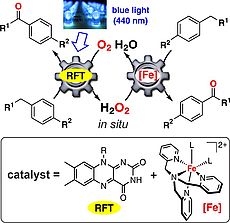
C-H Photooxygenation of Alkylbenzenes Catalyzed by Riboflavin Tetraacetate and a Non-Heme Iron Catalyst
The Sc(OTf)3/RFT system enables the oxygenation of alkylbenzenes with electron-withdrawing substituents, but this system still performs poorly for various other benzylic substrates. We developed a dual catalyst consisting of RFT and the tris(2-pyridylmethyl)amine complex [Fe(TPA)(MeCN)2](ClO4)2, which efficiently catalyzes the challenging photooxygenation of alkylbenzenes. The catalytic activity of the Fe complex for H2O2 disproportionation and alkylbenzene oxygenation contributes to the high efficiency of this catalyst combination.
.
.Angew. Chem. Int. Ed. 2016, 55, 427-430.
.
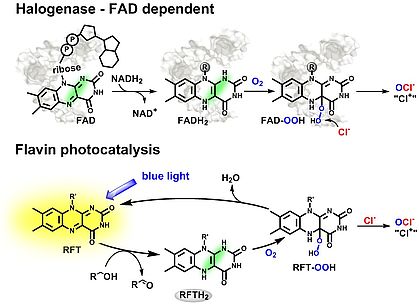
Halogenase-Inspired Oxidative Chlorination Using a Flavin Photocatalysis
In collaboration with group of Prof. Burkhard König (UR), we have developed a photocatalyst system for the selective chlorination of aromatic substrate. The RFT-based system is inspired by flavin adenine dinucleotide (FAD)-dependent halogenases. Compared to the high specificity of enzymes, our photocatalyzed protocol offers a more general strategy, allowing a broader substrate scope.
.
Angew. Chem. Int. Ed. 2016, 55, 5342-5345.
.
LOW-VALENT COMPLEXES
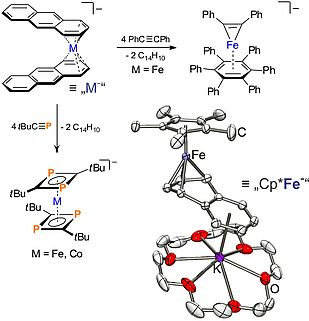
Low-valent Transition Metalate Anions
Low-valent alkene and polyarene complexes are used as sources of "Mx-"- and "CpMx-" synthons (M = transition metal, Cp = cyclopentadienyl ligand) for the synthesis of unusual new compounds and as new catalysts. Starting with anionic synthons, we synthesize "functional" anionic complexes, metal clusters and oligonuclear complexes. The catalytic properties are investigated in reductive transformations (see below). Our investigations focus on readily availabe and relatively cheap 3d metals, in particular iron and cobalt.
.
Chem. Commun. 2010, 46, 2832–2834; Dalton Trans. 2010, 39, 1453–1456; Chem. Eur. J. 2010, 16, 14322–14334; Inorg. Chem. 2012, 51, 6719–6730; Organometallics 2013, 32, 6040-6052; Dalton Trans. 2014, 43, 4247-4250.
.
Metalloradicals
We investigate the chemistry of new mononuclear 3d metal complexes with an open-shell electron configuration (metalloradicals). Our work currently focusses on the synthesis of new cyclopentadienyl metal(I) complexes. N-heterocyclic carbenes are used as ancillary ligands to stabilize these complexes in monomeric form. Their spectroscopic and magnetochemical characterization is carried out in collaboration with Prof. Bas de Bruin (University of Amsterdam) and Dr. Serhiy Demeshko (Prof. Franc Meyer, University of Göttingen).
NHC-stabilized cyclopentadienyl nickel(I) radicals are accessible in a straightforward manner by reducing nickel(II) halide complexes [CpNi(I)(NHC)Cl]. Spectroscopic investigations reveal that the these complexes can indeed be described as nickel-centered radicals.
.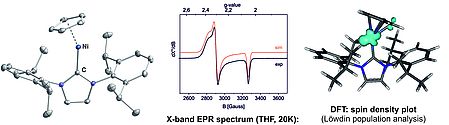
Chem. Commun. 2014, 50, 7014-7016.
White phosphorus is an excellent radical trap that reacts with these cyclopentadienyl nickel(I) compounds very selectively at low temperature. The resulting transition metal phosphides may serve as starting materials for the functionalization of the P4 moiety in the coordination sphere of the metal.
.
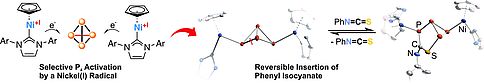
Chem. Commun. 2016, 52, 6601-6604..
In collaboration with Prof. Sjoerd Harder (University of Erlangen-Nürnberg) and Prof. Manfred Scheer (UR), we prepared a series of thermally stable half-sandwich complexes of the type [CpArM(μ-Br)]2. Taking the advantage of the bulky pentaarylcyclopentadienyl ligand C5(4-EtC6H4)5) (CpAr) and the distinct electronic properties compared to alkyl-substituted cyclopentadienyl ligands, these complexes may serve as a platform for new low valent transition metal chemistry. Employing [CpArNi(μ-Br)]2, a mononuclear cyclopentadienyl nickel(I) complex stabilized by a gallanediyl ligand Ga(nacnac) was synthesized. Spectroscopic and DFT studies show that the unpaired electron of this bimetallic Ni-Ga complex resides at the Ni center.
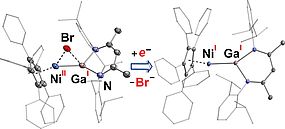
Inorg. Chem. 2016, 55, 3065-3074; Inorg. Chem. 2016, 55, 3075-3078.
Reduction of [CpArNi(μ-Br)]2 with KC8 in an aromatic solvent leads to a reactive "CpArNi" surrogate, which is trapped by N-heterocyclic carbenes, yellow sulfur and grey selenium, affording new Ni(I) and Ni(II) complexes.
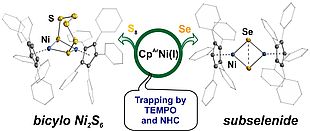
.
Organometallics 2016, 35, 1624-1631.
P-COMPOUNDS
.
Our research in organophosphorus and phosphaorganometallic chemistry is inspired by the diagonal relationship between phosphorus and carbon in the periodic table. Introducing P atoms into the scaffold of organic compounds and ligands significantly affects the electronic structures of the molecules, leading to changes in the properties of organometallic compounds. On-going projects specifically focus on phosphabenzenes and phosphaalkynes, which are used to synthesize compounds with fascinating new structures and properties.
.
Phosphatetrahedranes
In 2019, our group isolated di-tert-butyldiphosphatetrahedrane, (tBuCP)2, the first tetrahedral compound with phosphorus and carbon atoms in a tetrahedral scaffold. This rare ‘mixed’ tetrahedrane (tBuCP)2 is a hybrid of the P4 molecule and the purely carbon-based tetra-tert-butyltetrahedrane, tBu4C4. In addition, the synthesis of (tBuCP)2 is notable because it showed that a free phosphaalkyne dimer can be isolated as a stable species. (tBuCP)2 is synthesised by a facile nickel-catalyzed dimerization of tert-butylphosphaalkyne in yields of up to 55%, making it accessible for further reactivity studies which give rise to fascinating new organophosphorus and coordination compounds.
Very recent studies suggests that (tBuCP)2 is a versatile building block for organophosphorus chemisty. For example, (tBuCP)2 reacts with N-heterocyclic carbenes (NHCs) to afford unusual phosphaalkenes and phosphirenes. Moreover, the coordination chemistry of (tBuCP)2 is diverse: While Ag+ coordinates to the intact tetrahedron, nickel(I) complexes open P-C bonds of the P2C2 tetrahedron, forming new P/C ligand frameworks. Building on these initial results, further reactivity and coordination studies are on-going, which aim to unravel the full synthetic potential of this fascinating molecule and to target further phosphatetrahedranes.
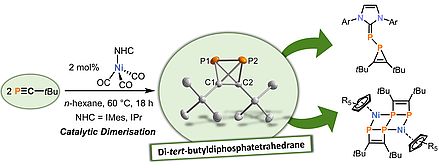
Angew. Chem. Int. Ed. 2019, 58, 16918–16922; Chem. Commun. 2021, 57, 2356–2359; Angew. Chem. Int. Ed. 2021, 60, 6435–6440.
.
Diphosphacyclobutadiene Sandwich Anions
Diphosphacyclobutadiene sandwich anions are conveniently accessible by reaction of phosphaalkynes with low-valent metalate anions. This program aims at the elucidation of reactivity and coordination properties of these phosphaorganometallic molecules and their use as synthetic building blocks for larger heterometallic aggregates. So far, these studies have already led to a range of hetero(bi)metallic complexes with unusual structural motifs and novel reactivity patterns.
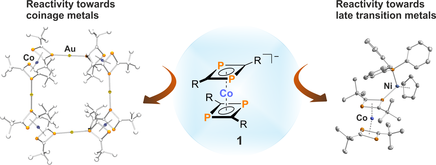
Eur. J. Inorg. Chem, 2016, 5, 736-742; Angew. Chem. Int. Ed. 2014, 53, 2771-2775.
.
Phosphinines: Coordination Chemistry, Small Molecule Activation and Catalytic Applications
Our group is interested in the synthesis, characterization and reactivity of phosphabenzenes (phosphinines), which are the heavier homologues of widely used pyridines and are fascinating due to their their versatile coordination chemistry and reactivity. Together with the group of Prof. Christian Müller (FU Berlin), we develop new coordination compounds of phosphinines with metalates. These complexes are able to activate a range of small molecules, including the unreactive CO2 molecule. Furthermore, we investigate the utility of phosphinine complexes and phosphacyclohexadienyl anions in homogeneous catalysis, e.g. in hydrofunctionalization reactions of alkenes, ketones and imines.
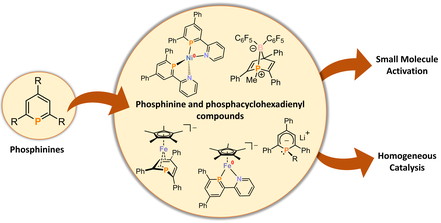
Organometallics 2015, 34, 622-635; Dalton Trans. 2016, 45, 8875–8884; Eur. J. Inorg. Chem. 2019, 1567–1574; Chem. Eur. J. 2020, 26, 7788–7800; ACIE. 2019, 58, 15407–15411; Inorg. Chem. 2020, 59, 9951–9961.

